Blog
Vigilanzsystem gemäß (EU) 2017/745
Meldepflicht gemäß der MDR
Wird ein Vorkommnis nach der ersten Bewertung nicht als schwerwiegendes Vorkommnis eingestuft, muss dennoch geprüft werden, ob es zu einem der in Artikel 2 (65) a-c der MDR genannten Ergebnisse führen könnte, wenn die Umstände weniger günstig wären (z. B. ohne die Intervention eines Dritten oder wenn mehr gefährdete Patienten derselben Situation ausgesetzt gewesen wären usw.).
Wenn der Hersteller nicht ausschließen kann, dass das Vorkommnis möglicherweise zu den in Artikel 2 (65) a- c MDR genannten Folgen geführt haben könnte, ist das Vorkommnis als schwerwiegend zu betrachten und muss der zuständigen Behörde gemeldet werden. Ist sich der Hersteller nach Bekanntwerden eines möglicherweise meldepflichtigen Vorkommnisses unsicher darüber, ob das Vorkommnis meldepflichtig ist, muss er dennoch innerhalb der in Artikel 87(2) gemäß Artikel 87 (2-5) MDR einen Bericht vorlegen.
Das nachstehende Flussdiagramm veranschaulicht den von den Herstellern zu befolgenden Prozess für das Management von Vorkommnissen und schwerwiegenden Vorkommnissen.
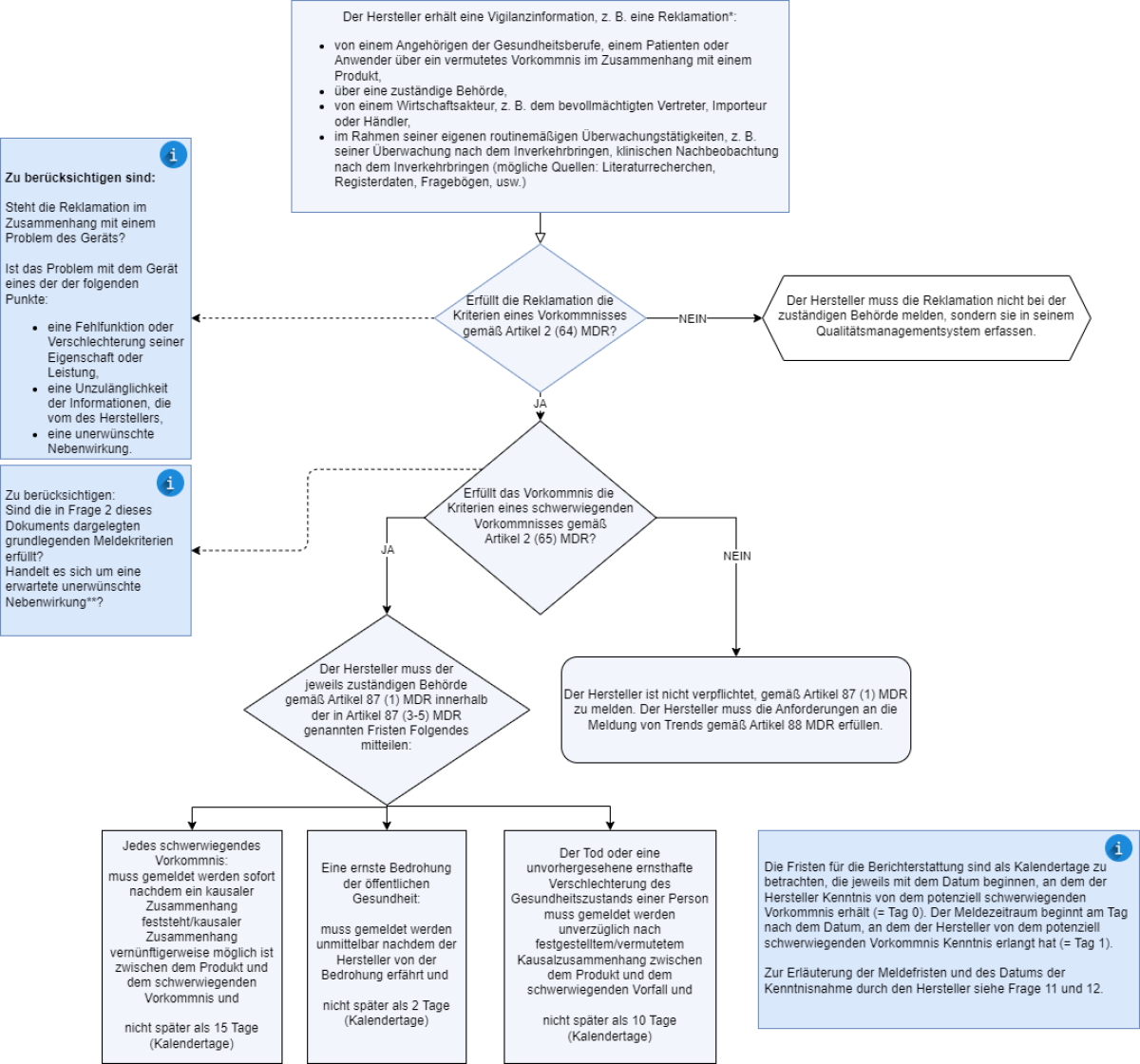
Bilderquelle: Leitfaden MDCG 2023-3 “Questions and Answers on vigilance terms and concepts as outlined in the Regulation (EU) 2017/745 on medical devices”, Seite 4.
Was versteht man unter einer Reklamation?
Eine Reklamation ist in der EN ISO 13485:2016 definiert und kann als schriftliche, elektronische oder mündliche Mitteilung beschrieben werden, die sich auf angebliche Unzulänglichkeiten in Bezug auf die Identität, Qualität, Haltbarkeit, Zuverlässigkeit, Gebrauchstauglichkeit, Sicherheit oder Leistung eines Medizinprodukts oder im Zusammenhang mit einer Dienstleistung, die die Leistung eines solchen Medizinprodukts beeinträchtigt. Es ist wichtig zu beachten, dass Reklamationen und Informationen, die für die Vigilanzmeldung relevant sind, nicht nur von außen kommen, sondern auch aufgrund der eigenen Aktivitäten des Herstellers zur routinemäßigen Überwachung der Sicherheit und Leistung eines Produkts entstehen können.
Was versteht man unter unerwünschte Nebenwirkungen?
Unter einer “unerwünschten Nebenwirkung” im Sinne der MDR ist jede unbeabsichtigte und unerwünschte medizinische Erscheinung im menschlichen Körper, die sich aus der normalen Anwendung eines Produkts ergibt. Unerwünschte Nebenwirkungen sind nicht das Ergebnis einer Fehlfunktion, einer Verschlechterung der Eigenschaften oder der Leistung des Produkts oder einer unzureichenden Information durch den Hersteller. Eine erfolglose Behandlung (oder ein Behandlungsfehler) sollte nicht als unerwünschte Nebenwirkung angesehen werden. Für die Zwecke dieses Leitfadens können unerwünschte Nebenwirkungen erwartet oder unerwartet sein und werden als Zwischenfälle im Sinne der MDR betrachtet (Artikel 2(64) MDR).
Erwartete unerwünschte Nebenwirkungen werden in der Produktinformation klar dokumentiert, in der technischen Dokumentation quantifiziert und unterliegen der Meldung von Trends gemäß Artikel 88 MDR.
Der Unterschied zwischen Vorkommnis und schwerwiegendes Vorkommnis
„Vorkommnis“ bezeichnet eine Fehlfunktion oder Verschlechterung der Eigenschaften oder Leistung eines bereits auf dem Markt bereitgestellten Produkts, einschließlich Anwendungsfehlern aufgrund ergonomischer Merkmale, sowie eine Unzulänglichkeit der vom Hersteller bereitgestellten Informationen oder eine unerwünschte Nebenwirkung.
„Schwerwiegendes Vorkommnis“ bezeichnet ein Vorkommnis, das direkt oder indirekt eine der nachstehenden Folgen hatte, hätte haben können oder haben könnte:
- den Tod eines Patienten, Anwenders oder einer anderen Person
- die vorübergehende oder dauerhafte schwerwiegende Verschlechterung des Gesundheitszustands eines Patienten, Anwenders oder anderer Personen
- eine schwerwiegende Gefahr für die öffentliche Gesundheit
Nähere Informationen zu Begriffen und Konzepte der Vigilanz finden Sie im folgenden MDCG Leitfaden: Leitfaden MDCG 2023-3
Medizinprodukte der Risikoklasse I
Wir schaffen Klarheit
Leider kam es in jüngster Vergangenheit immer wieder zu Missverständnissen bezüglich der zu tätigenden Dokumentationen und Zertifizierungen eines reinen Klasse I Medizinproduktes. Wir fassen die wichtigsten Eckpunkte für Sie zusammen.
Ein reines Klasse I Medizinprodukt…
- … muss den Nachweis der relevanten grundlegenden Sicherheits- und Leistungsanforderungen erbringen.
- MDR, Annex III
- … benötigt eine technische Dokumentation.
- MDR, Annex II + Annex III
Für ein reines Klasse I Medizinprodukt…
- … muss eine klinische Bewertung durchgeführt werden.
- … muss - sofern die klinischen Daten nicht ausreichend sind - eine klinische Studie aufgesetzt werden.
- … hat der Hersteller selbst die Konformitätserklärung auszustellen
- … benötigt der Hersteller für die Zulassung keine Benannte Stelle
Der Medizinprodukte-Hersteller eines solchen sogenannten “Niedrig-Risiko-Produktes” hat ein QMS (Qualtitätsmanagement-System, siehe MDR, Art. 10) zu etablieren. Dieses 13485-QMS muss in diesem Fall jedoch nicht zwangsweise zertifiziert sein bzw. werden!
Die angeführte Grafik liefert die aus dem MDCG-Dokument “MDCG Guidance for Class I Medical Device Manufaturers” zu entnehmenden wichtigsten Schritte, die Hersteller bei der Zulassung und dem Aufbau der dafür benötigten Dokumention zu beachten haben.

QMD Services - Die einzige Benannte Stelle Österreichs
Österreich hat eine neue Benannte Stelle für IVD Produkte! Wir haben die näheren Informationen für Sie!

Was bietet QMD Services aktuell?
- Konformitätsbewertung nach IX und XI
- Zertifizierung nach Artikel 16 (3) IVDR
- Importeure oder Händler, die Produktkennzeichnungen übersetzten oder (nicht sterile) IVD umpacken oder vereinzeln, benötigen:
- QMS
- IVDR Art. 16 (3) Zertifikat
- ISO 13485 Zertifizierung (mit Quality Austria)
- Weitere Scopes für IVDR sind zu erwarten
- Akkredierung nach MDR noch ausständig
Aktuelle QMD Services IVDR Codes (Stand: 31.01.23):
| Code | Beschreibung |
|---|---|
| 0301-0302 | 3. Produkte für Marker für Krebs und gutartige Tumoren |
| 0401-0403 | 4. Produkte zur Durchführung von Gentests beim Menschen |
| 0501-0506 | 5. Produkte zum Bestimmung der Marker von Infektionen/zur Bestimmung des Immunstatus |
| 0601-0609 | 6. Produkte für nicht infektiöse Krankheiten, physiologische Marker, Störungen/Beeinträchtigungen (außer der Durchführung von Gentests beim Menschen) und für therapeutische Maßnahmen |
Die Codes für das Design und die Zweckbestimmung, als auch weiter horizontale Codes sind unter folgendem Link ersichtlich: Scope Liste
Was ist für die Einreichung der Technische Dokumentation essenziell?
- Vollständige oder fertigwerdende Technische Dokumentation
- Wenn Bereiche noch nicht vollständig sind (z. B. Stabilitätstests) Begründung für die Nachreichung
- Vor der Einreichung des Antrags sollten Hersteller mit der Benannten Stelle über die notwendige Struktur sprechen (prinzipielle Orientierung nach IVDR, Anhang II)
- In der Technischen Dokumentation müssen Herstellerangaben enthalten sein: Name, Anschrift, EMDN-Codierung und NBOG-Codierung und ggf. Sitz EU-Bevollmächtigten, kritischen Lieferanten und/oder Unterauftragnehmer
- Sprache der Technischen Dokumentation, sollte grundsätzlich in EN sein.
- Bei manchen Scopes kann es Ausnahmen geben und die Technische Dokumentation kann in deutscher Sprache verfasst werden (wir empfehlen hierbei jedoch, noch in sehr früher Entwicklungsphase Rücksprache mit QMD Services zu halten).
- Ggf. sollten zuletzt aktualisierte umfassende Berichte und Daten beigefügt werden
- Gekürzte und unvollständige Prüfberichte werden nicht akzeptiert
- Prüfberichte müssen vollständig sein (kein Bericht mit nachträglichen Änderungen oder Revisionen - da Produkt verändert wurde)
- Unterlagen müssen dokumentieren, wie der Hersteller die Einhaltung aller geltenden GSPR sicherstellt
- Informationen in allen Bereichen (in denen sich Informationen wiederholen) müssen korrekt sein
- Aktualisierung: Risiko berücksichtigen von potenziellen Fehlern / Inkonsistenzen (zB. UDI-DI, Verwendungszweck, Anwendungshinweise, Kontraindikationen, Warnhinweise…)
- Wenn die angeforderten Daten unzureichend sind, sollten stets stichhaltige Begründungen vorgelegt oder beigefügt werden.
- Hersteller hält Liste aller von ihm vergebenen UDIs auf dem neuesten Stand (gem. Anforderung Anhang II IVDR)
Grobe Beschreibung Ablauf Bewerbung:
- Online Kontaktformular ausfüllen (Rückmeldung erfolgt zurzeit innerhalb von ca. 2 Tagen)
- Virtuelles Meeting
- Zusendung von Produkt & Firmen Informationen seitens Hersteller
- Übermittlung der Kostenschätzung seitens QMD Services
- Hersteller können sich einen ersten eigenen Überblick über die Kosten machen: Kosten
- Rücksendung der Kostenschätzung inkl. Unterschrift
- Angebot Zusendung seitens QMD Services
- Rücksendung Angebot (inkl. Unterschrift)
- Vertrag Zusendung seitens QMD Services
- Rücksendung Vertrag (inkl. Unterschrift)
- START KONFORMITÄTSBEWERTUNGSVERFAHREN
Detailreicher Ablauf: Bewerbungsablauf
Ursachen für lange Konformitätsbewertungsverfahren:
- Unvollständige Einreichungen - fehlende Informationen
- Fehlende kohärente Struktur der Technischen Dokumentation / schwer auffindbare Informationen
Zukünftig:
- Es werden seitens QMD Services künftig Schulungen für die Erstellung der Technischen Dokumentation - laut IVDR - geplant
- Es wird eine MDCG Leitlinie erstellt werden (Best Practice Guideline für die IVDR Technische Dokumentation)
Quelle: Online Inforveranstaltung QMD Services am 25.01.23
RnB-Consulting unterstützt Sie im Rahmen der Implementierung der europäischen Verordnung für Medizinprodukte/In vitro Diagnostika (MDR/IVDR) und steht dabei mit erfahrenen Experten gerne zur Seite. Schicken Sie Ihre unverbindliche Anfrage an: office@rnb-consulting.at
Übergangsverordnung_NEU
Wenn der Drang nach mehr Sicherheit dazu führt, dass (lebens)wichtige Medizinprodukte nicht für Patient*innen zur Verfügung stehen… 😕
 Die aktuelle Änderung hinsichtlich der Übergangsverordnung für bestimmte Medizinprodukte und In-vitro-Diagnostika - nach (EU) 2017/745 und (EU) 2017/746, veranschaulicht die gegenwärtige Problematik:
Von zur Zeit 21.376 gültigen Zertifikaten nach RL 90/385/EWG (AIMD) für aktiv-implantierbare Medizinprodukte & RL 93/42/EWG (MDD) für allgemeine Medizinprodukte, laufen 4.311 im Jahr 2023 und 17.095 in den ersten 5 Monaten des Jahres 2024 aus.
Die aktuelle Änderung hinsichtlich der Übergangsverordnung für bestimmte Medizinprodukte und In-vitro-Diagnostika - nach (EU) 2017/745 und (EU) 2017/746, veranschaulicht die gegenwärtige Problematik:
Von zur Zeit 21.376 gültigen Zertifikaten nach RL 90/385/EWG (AIMD) für aktiv-implantierbare Medizinprodukte & RL 93/42/EWG (MDD) für allgemeine Medizinprodukte, laufen 4.311 im Jahr 2023 und 17.095 in den ersten 5 Monaten des Jahres 2024 aus.
Was dann? 😦
Gibt es überhaupt genügend Kapazitäten der aktuell 36 Benannten Stellen, um diesen kommenden Workload bändigen zu können? Was ist mit neuen Innovationen - Neuzertifizierungen? Müssen diese dann “hintenangestellt” werden? Was bedeutet dies dann für Start-ups? Wer überwacht jene Medizinprodukte mit abgelaufenem Zertifkat, die sich weiterhin in Verwendung befinden?
Das übergeordnete Ziel der vorgeschlagenen Änderungen besteht darin, den Zugang der Patient*innen zu einer großen Auswahl an Medizinprodukten bei gleichzeitiger Sicherstellung des Übergangs zum neuen Rahmenwerk zu gewährleisten. Die Verlängerung erfolgt je nach Risikoklasse des Gerätes gestaffelt, d.h. bis Dezember 2027 für Geräte mit höherem Risiko und bis Dezember 2028 für mittlere und geringerem Risiko. Dies würde Herstellern und benannten Stellen mehr Zeit für die Durchführung der Konformitätsbewertungsverfahren gemäß der MDR geben.
Zudem wird vorgeschlagen, die “Abverkaufsfrist” zu streichen. Also jenes Datum, nach dem in Verkehr gebrachte Produkte vom Markt genommen werden müssen. Dies würde eine unnötige Entsorgung sicherer Medizinprodukte verhindern.
Lesen Sie mehr zu den vorgeschlagenen Änderungen der Übergangsfirsten unter: https://health.ec.europa.eu/system/files/2023-01/mdr_proposal.pdf
Sie haben Fragen dazu und/oder unseren Services? Gerne unter: office@rnb-consulting.at
Aufbau der Technischen Dokumentation für IVDs - Welche Auswirkung haben die Risikoklassen darauf?
IVDs sind in folgende Klassen unterteilt:
| Risikoklasse | Beispiele |
|---|---|
| D | HIV, Syphilis |
| C | HLA, Rubella |
| B | Blutgase, physiologische Marker: Hormone |
| A | Pufferlösungen, Probenbehältnisse |
Aktuell begleiten wir den Aufbau der Technischen Dokumentation für ein Klasse A IVD. Auch wenn in diese IVD-Klasse unkritische Produkte wie bspw. Probenbehältnisse oder Instrumente für die Verwendung bei In-vitro Diagnoseverfahren fallen, ist die Einhaltung der Grundlegenden Sicherheits- und Leistungsanforderungen (IVDR, Anhang I) obligatorisch.
In diesem Zusammenhang ist eine wichtige Information zu erwähnen: IVD-Hersteller aller IVD-Klassen sind somit verpflichtet:
- Ein QM aufzubauen
- Die Gebrauchstauglichkeit nach EN 62366 nachzuweisen
- Die direkte Verbindung zum Risikomanagement nach EN 14971 herzustellen
- Verifizierungen und Validierungen des Produktes nachzuweisen
Was jedoch den größeren Unterschied der IVD-Klassen untereinander auszeichnet, ist der zu wählende Zertifizierungsprozess. Reine Klasse A IVDs benötigen keine BS (Benannte Stelle) und Hersteller dieser IVD-Klassen, können die Konformitätserklärung eigenständig ausstellen.
Bei In-vitro Diagnostika der Klasse A steril wird seitens des Herstellers eine Technische Dokumentation erstellt und Teilbereiche des Qualitätsmangementsystem seitens der Benannten Stelle geprüft.
Bei Klasse B und C IVDs ist die Benannte Stelle dazu verpflichtet, das Qualitätsmanagementsystem und die Technische Dokumentation zu prüfen.
Bei Klasse D IVDs wird eine detailreiche Auditierung durchgeführt. Das Referenzlaboratorium (Artikel 100 IVDR) ist in die Prüfungen miteinbezogen (Überprüfung der vom Hersteller angegebenen Leistung und ggf. der Einhaltung der Gemeinsamen Spezifikationen). Gemäß Artikel 50 IVDR kann die Koordinierungsgruppe Medizinprodukte und ggf. Kommission bei begründeten Bedenken die Expertengremien miteinbeziehen, um wissenschaftliche Gutachten zur Sicherheit und Leistung von Produkten zu erhalten.
Nähere Informationen zu den Konformitätsbewertungsverfahren finden Sie im foglenden Bild:
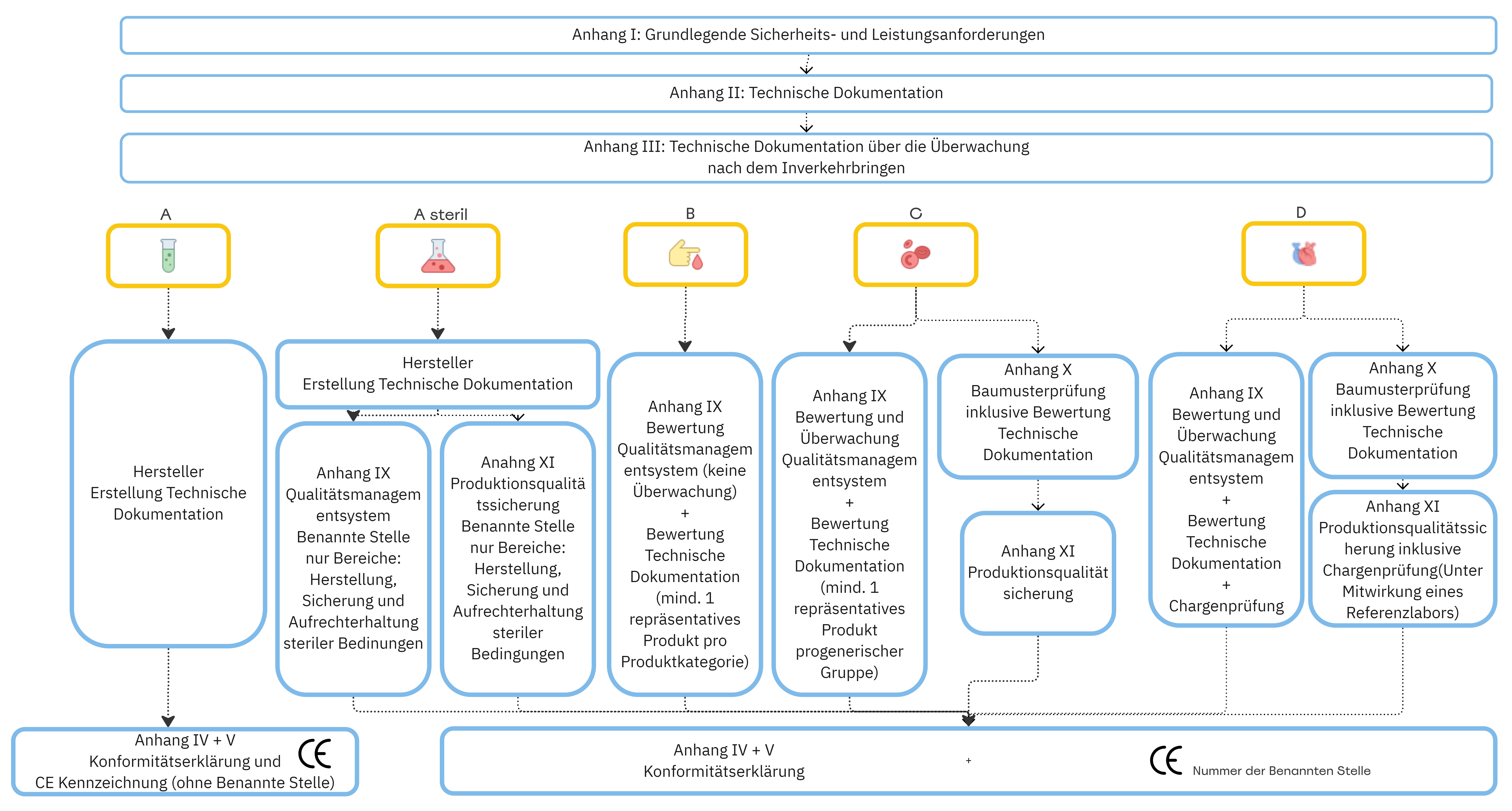
Bildquelle: TÜV SÜD Seminar In-vitro-Diagnostika - Inhalte und Umsetzung der neuen europäischen IVD-Verordnung