Blog
Was ist der PSUR, was fordert dieser von Medizinprodukte-Herstellern und welche Unterschiede gibt es bzgl. Risikoklassen?

Eine der neuen Anforderungen, die mit der EU-MDR 2017/745 eingeführt wurden, betrifft die Erstellung des regelmäßig aktualisierten Berichtes über die Sicherheit (engl. PSUR). Der regelmäßig aktualisierte Bericht über die Sicherheit (engl. Periodic safety update report - PSUR), ist ein Dokument, das die Überwachungsdaten des Produkts nach dem Inverkehrbringen zusammenfasst.
Wie Sie in der unteren Tabelle sehen können, ist der PSUR ab der Risikoklassen IIa bis III obligatorisch.
| Risikoklasse | PSUR |
|---|---|
| I | Nein |
| IIa | Ja |
| IIb | Ja |
| III | Ja |
Bei Produkten der Klasse I wird der regelmäßige aktualisierte Bericht über die Sicherheit durch einen Bericht über die Überwachung nach dem Inverkehrbringen (engl. PMS-Report) ersetzt. Aufgrund dessen, dass es sich beim PMS-Report um niedrig klassifizierte Produkte handelt (Klasse I), enthält er auch weniger umfangreiche Informationen.
Was sind die Inhalte des PSURs?
Die Anforderungen an den regelmäßig aktualisierten Sicherheitsbericht sind in Artikel 86 der EU MDR 2017/745 festgelegt. Der PSUR wird für jedes Medizinprodukt erstellt. Im Einzelnen muss der PSUR folgendes enthalten:
- die Schlussfolgerung der Nutzen-Risiko-Abwägung
- die wichtigsten Ergebnisse der klinischen Nachbeobachtung nach dem Inverkehrbringen (engl. post-market clinical follow-up - PMCF)
- das Verkaufsvolumen des Produkts und andere Merkmale der Bevölkerungsgruppe, die das Produkt verwendet, sowie, soweit durchführbar, die Häufigkeit der Verwendung des Produkts.
Der Sicherheitsbericht enthält die Ergebnisse und Schlussfolgerungen der Analysedaten aus der Überwachung nach dem Inverkehrbringen (engl. post-market surveillance data), die im Rahmen des Plans zur Überwachung nach dem Inverkehrbringen (engl. post-market surveillance plan) gesammelt wurden, sowie eine Begründung und Beschreibung aller ergriffenen Präventions- und Korrekturmaßnahmen.
Wie oft muss ein regelmäßiger aktualisierter Bericht über die Sicherheit dokumentiert werden?
Die Verpflichtung zur Erstellung des PSUR beginnt mit dem Datum der Anwendung der MDR. Ein Update des PSUR ist abhängig von der Risikoklasse des Medizinproduktes. Insbesondere für Medizinprodukte der Klasse IIa ist der PSUR alle zwei Jahre zu erstellen und wird von der benannten Stelle im Rahmen der Konformitätsbewertung überprüft. Für Medizinprodukte der Klasse IIb oder der Klasse III ist der PSUR mindestens einmal jährlich zu dokumentieren. Die Erstellung von PSURs sollte während der gesamten Lebensdauer des Produkts fortgesetzt werden.
Wer soll den PSUR überprüfen?
Gemäß der EU MDR 2017/745 muss die Benannte Stelle die PSURs für Produkte der Klasse III und implantierbare Produkte jährlich überprüfen. Bei Produkten der Klassen IIa und IIb wird die Prüfung im Rahmen des Überwachungsaudits durchgeführt. Die zuständigen Behörden können PSURs auch im Rahmen ihrer Vigilanzuntersuchungen, Überprüfungen klinischer Prüfungen und Marktüberwachungstätigkeiten anfordern und überprüfen.
Quelle: QualityMedDev; 17.10.22
Übergangsfristen und Technische Dokumentation nach IVDR
Was für die unterschiedlichen Risikoklassen der IVD Produkte zu beachten ist
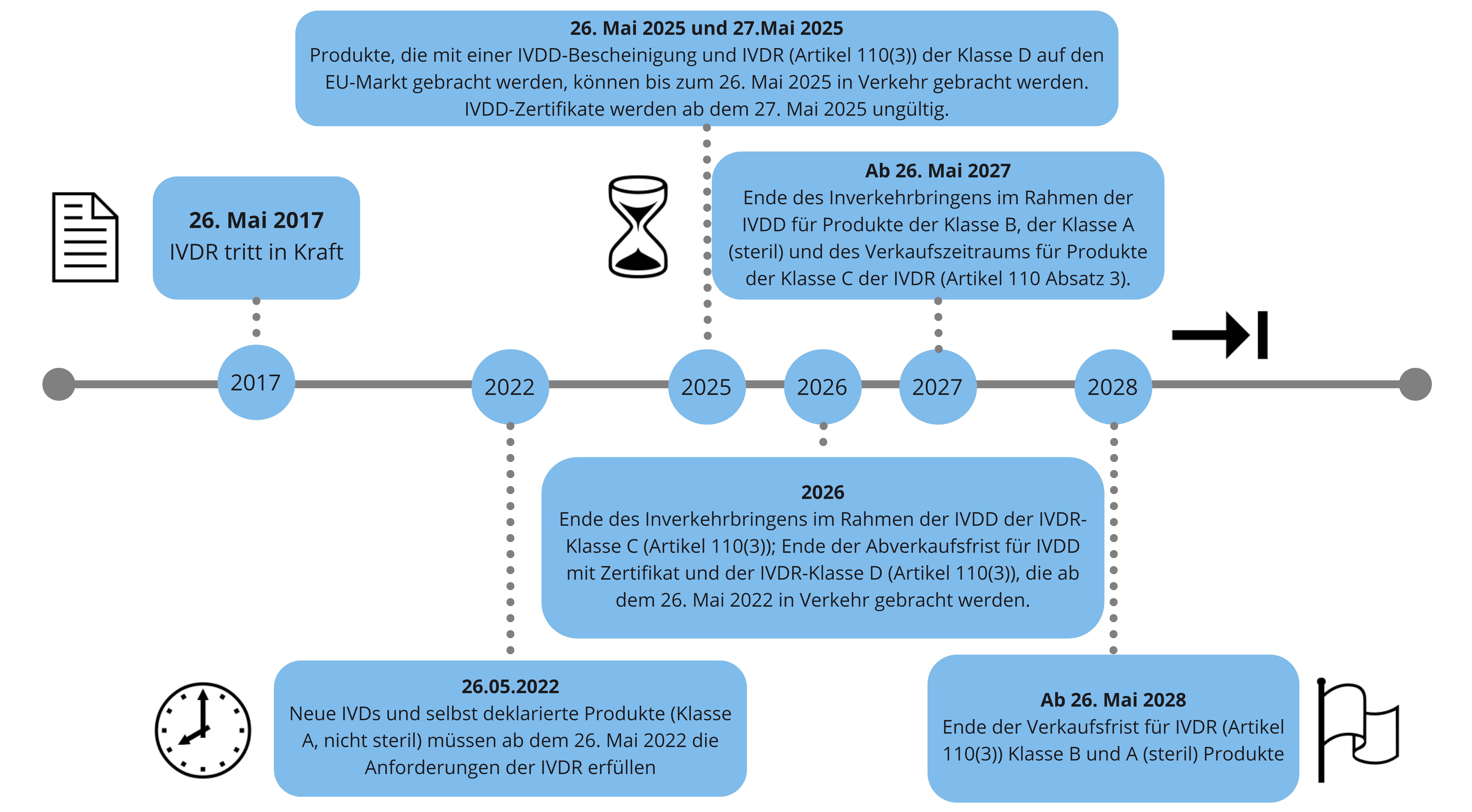
Die Übergangsfristen sind in Artikel 110 der IVDR festgelegt – wir möchten diese für Sie zusammenfassen und visualisieren.
Die IVDR (engl.: In vitro Diagnostic Regulation) ist am 05. Mai 2017 im EU-Amtsblatt bekannt gemacht worden und am 26. Mai 2017 in Kraft getreten. Die Übergangsfrist für die IVDR ist auf 5 Jahre gesetzt worden, somit gilt diese ab 26.Mai 2022. Die Pandemie veranlasste die EU-Kommission jedoch dazu, die speziellen Übergangsbestimmungen anzupassen - die Verordnung (Verordnung 2022/112) dahingehend, wurde am 28. Jänner 2022 veröffentlicht.
Für ein IVD mit gültigem IVDD-Zertifikat, welches vor dem 26. Mai 2022 ausgestellt wurde, gilt Artikel 120 Absatz 3 w,elcher auf sogenannte Bestandsprodukte abzielt.
Produkte, für die gemäß der Richtlinie 98/79/EG eine aufgrund von Absatz 2 gültige Bescheinigung ausgestellt wurde, dürfen bis zum 26. Mai 2025 in Verkehr gebracht oder in Betrieb genommen werden. (Achtung: sofern keine wesentlichen Änderungen der Auslegung und Zweckbestimmung vorliegen!)
Hinweis: Bitte achten Sie bei Ihrem Projektmanagement bzw. der zeitlichen Planung Ihrer Zulassung, auf die derzeitige Auslastung der Benannten Stellen. Ist für Ihr In-vitro Diagnostikum eine Konformitätsbewertung durch eine Benannte Stelle notwendig, sorgen Sie frühzeitig dafür dementsprechende Angebote und Informationen hinsichtlich Auslastung einzuholen.
Produkte, für die das Konformitätsbewertungsverfahren gemäß der Richtlinie 98/79/EG nicht die Mitwirkung einer benannten Stelle erforderte, für die vor dem 26. Mai 2022 eine Konformitätserklärung gemäß der genannten Richtlinie ausgestellt wurde und für die das Konformitätsbewertungsverfahren gemäß der vorliegenden Verordnung die Mitwirkung einer benannten Stelle erfordert, dürfen bis zu folgenden Zeitpunkten in Verkehr gebracht oder in Betrieb genommen werden:
| Zeitpunkt | Risikoklasse |
|---|---|
| 26. Mai 2025 | Klasse D |
| 26. Mai 2026 | Klasse C |
| 26. Mai 2027 | Klasse B |
| 26. Mai 2027 | Klasse A (in sterilem Zustand in Verkehr gebracht) |
Sie haben Fragen zu diesem Blog, anderen Themen oder zu unserem Unternehmen?
Schicken Sie Ihre unverbindliche Anfrage an: office@rnb-consulting.at
Übergangsfristen der technische Dokumentation nach MDR
Was für die unterschiedlichen Risikoklassen der Medizinprodukte zu beachten ist
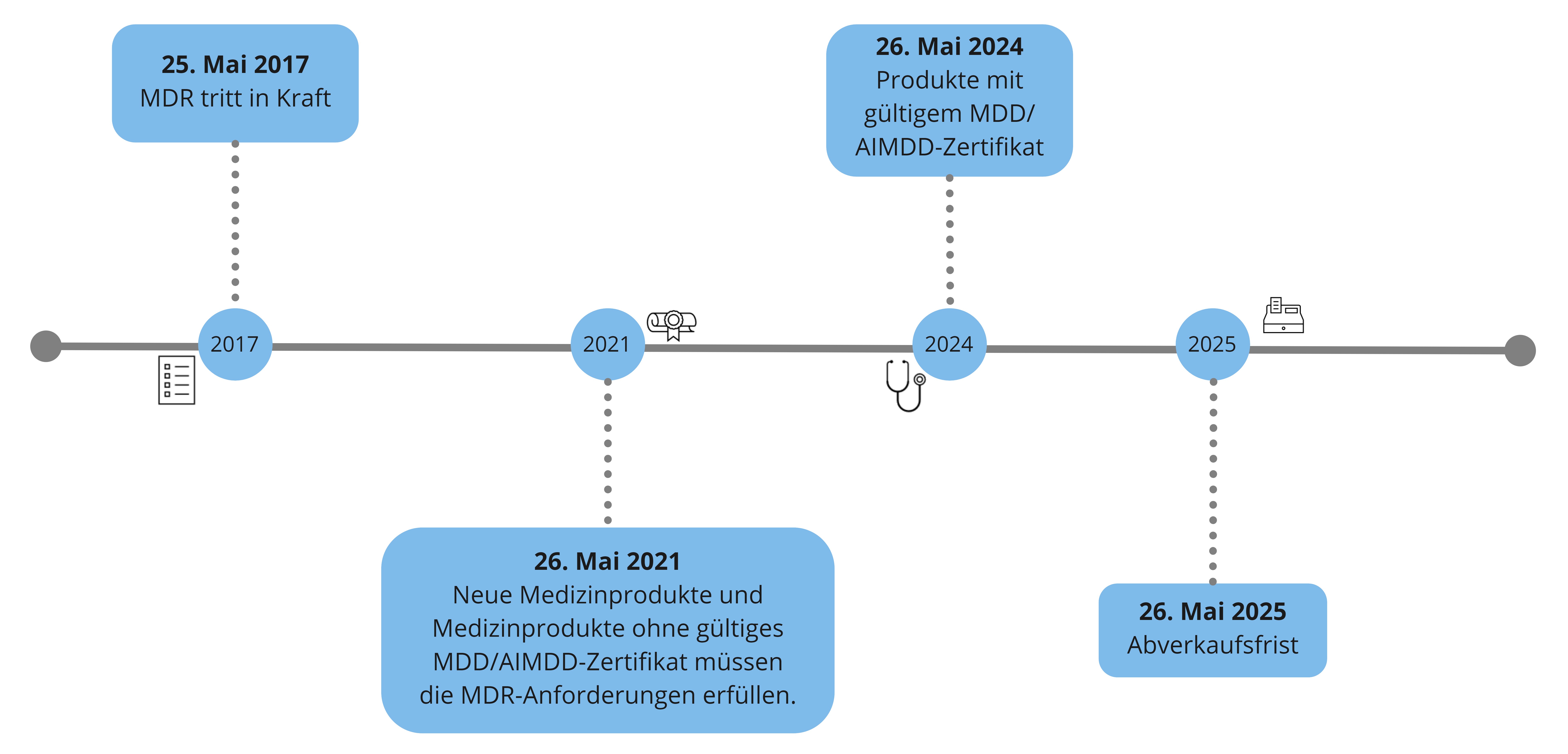
Die Übergangsfristen sind in Artikel 120 der MDR festgelegt – wir möchten diese für Sie zusammenfassen und visualisieren.
Die MDR (engl.: Medical Device Regulation) ist am 05. Mai 2017 im EU-Amtsblatt bekannt gemacht worden und am 25. Mai 2017 in Kraft getreten.
Die Übergangsfrist für die MDR war anfänglich auf 3 Jahre gesetzt worden.
Die Pandemie veranlasste die EU-Kommission dazu, diese auf 4 Jahre auszuweiten - die Verordnung (Verordnung 2020/561) dahingehend, wurde am 24. April 2020 veröffentlicht.
Medizinprodukte mit gültigem MDD/AIMDD-Zertifikat, welches vor dem 26. Mai 2021 ausgestellt wurde, gilt Art. 120, Absatz 3, welcher auf sogenannte Bestandsprodukte abzielt. Diese Produkte dürfen bis zum 26. Mai 2024 in Verkehr gebracht oder in Betrieb genommen werden. (Achtung: sofern keine wesentlichen Änderungen der Auslegung und Zweckbestimmung vorliegen!)
Bis 26. Mai 2025 ist die Bereitstellung auf dem Markt und Inbetriebnahme dieser Medizinprodukte möglich (=Abverkaufsfrist).
Hinweis: Bitte achten Sie bei Ihrem Projektmanagement bzw. der zeitlichen Planung Ihrer Zulassung, auf die derzeitige Auslastung der Benannten Stellen. Ist für Ihr Medizinprodukt eine Konformitätsbewertung durch eine Benannte Stelle notwendig, sorgen Sie frühzeitig dafür dementsprechende Angebote und Informationen hinsichtlich Auslastung einzuholen.
Welchen Einfluss hat die Risikoklasse eines Medizinproduktes auf die in Annex II und Annex III der MDR geforderten technischen Dokumentation?
Medizinprodukte sind in unterschiedliche Risikoklassen (I, Ir, Im, Is, IIa, IIb, III) einzustufen. Diese Einstufung erfolgt anhand sogenannter Klassifizierungsregeln (MDR, Annex VIII), basierend auf der vorabdefinierten Zweckbestimmung des Medizinproduktes.
Je nach Risikoklasse - haben Sie als Medizinprodukte-Hersteller – ein Konformitätsbewertungsverfahren zu wählen:
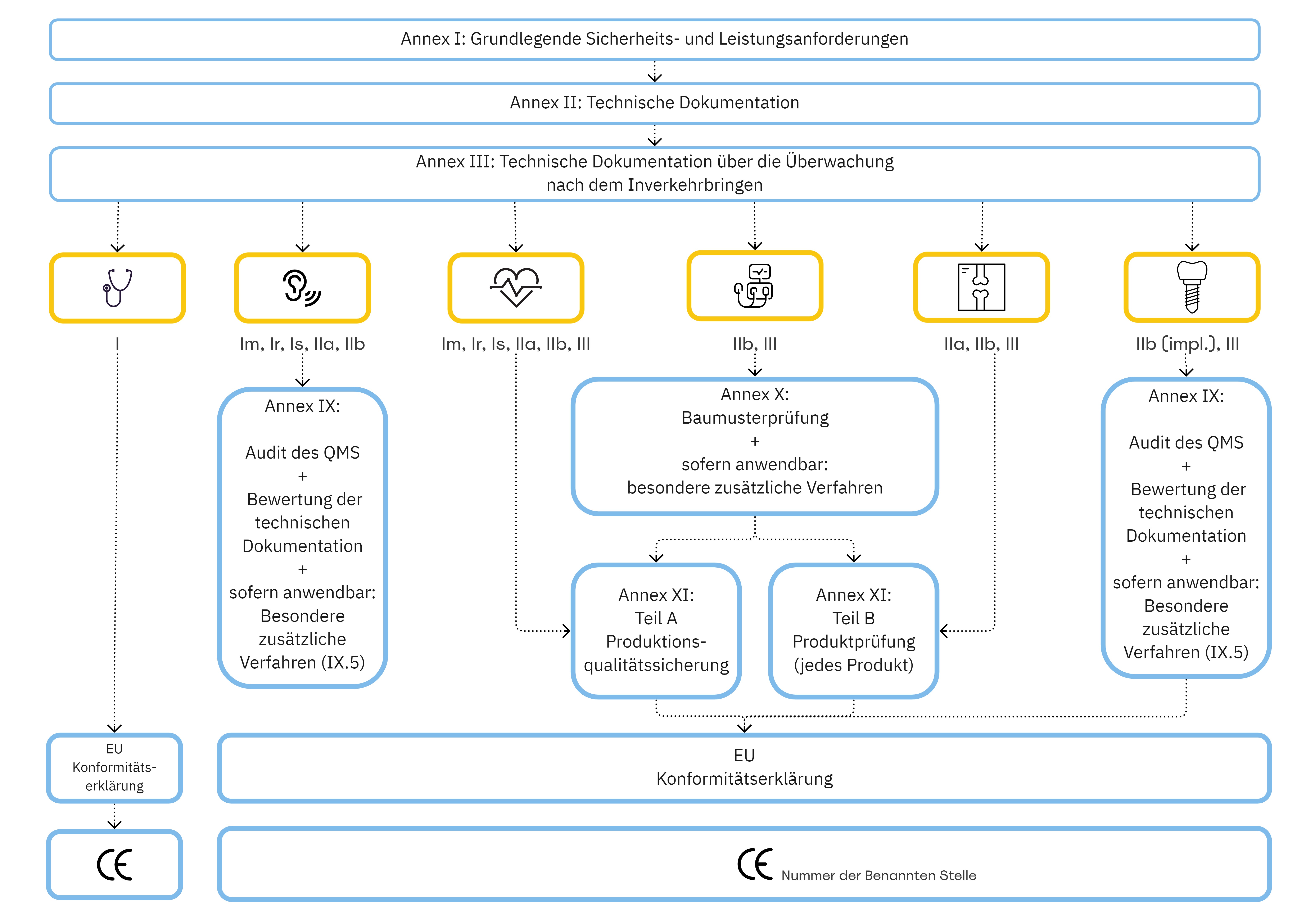
Die technische Dokumentation ist für Medizinprodukte – egal welche Risikoklasse diese aufweisen – zu erstellen. Vor allem der Nachweis der grundlegenden Sicherheits- und Leistungsanforderungen (MDR, Annex I) ist essenziell für eine erfolgreiche Zertifizierung. Der Nachweis dieses Annex I geht mit der Erstellung der technischen Dokumentation einher, da diese den dokumentierten Nachweis der Einhaltung der einzelnen Anforderungen repräsentiert.
Sie haben Fragen zu diesem Blog, anderen Themen oder zu unserem Unternehmen? Schicken Sie Ihre unverbindliche Anfrage an: office@rnb-consulting.at
RnB-Workshops – Start im Herbst 2022!
Freuen Sie sich auf spannende und vor allem individuell auf Ihre Bedürfnisse angepasste Workshoptage!
Was wir NICHT wollen:
- Frontalvorträge
- Theoretische Erklärungen
- Keinen Mehrwert für Sie und Ihr Unternehmen
Was wir WOLLEN:
- Spezifische Workshops - angepasst an Sie, Ihr Unternehmen, Ihr(e) Medizinprodukt(e)
- Beispiele aus der Praxis
- Ihnen einen Fahrplan und bereits erste Dokumentenentwürfe für Ihre Umsetzungen mit auf den Weg geben
Unsere Themen im Überblick:
- Risikomanagement
- Usability Engineering
- Klinische Bewertung / Klinische Prüfung
- MDR-Umsetzung
- IVDR-Umsetzung
- Software-Entwicklung
- Projektmanagement
- Qualitätsmanagement
- Produktentwicklung
- Post Market Surveillance
Unsere Workshops – individuell an Sie angepasst:
Nach einem kurzen Ein- und Überblick über das aktuell geltende regulatorische Rahmenwerk der Medizintechnik, gehen wir gemeinsam auf Praxisbeispiele und Erfahrungen Ihrerseits ein.
Den größten Part des Workshops umfasst neben der Erläuterung, wie Sie geforderte Dokumentationen umsetzen, die Erstellung eines für Sie angepassten „Fahrplans“.
Dieser bringt Ihnen einen großen Mehrwert, da Sie damit nach dem RnB-Workshop gut ausgerüstet sind, um den regulatorischen Anforderungen für Ihr(e) Medizinprodukt(e) gerecht zu werden.

Sie haben Fragen rund um unsere RnB-Workshops? Kontaktieren Sie uns unter: office@rnb-consulting.at
Usability Engineering - angepasst an Ihr Medizinprodukt
Der Alltag von Gesundheitsdienstleistern ist geprägt von der Anwendung innovativer Medizinprodukte. Auch in den eigenen vier Wänden nehmen immer mehr medizintechnische Gerätschaften einen fixen Platz ein und werden in das alltägliche Leben integriert.
 Eine unzureichende Gebrauchstauglichkeit dieser Medizinprodukte kann oftmals schwerwiegende Konsequenzen mit sich tragen, Anwenderfehler können zu schlimmen Verletzungen und – im WorstCase – gar zum Tode führen.
Medizinprodukte sollten uns eine Hilfestellung geben, die Lebensqualität zu verbessern, Untersuchungsprozesse zu optimieren und die bestmögliche medizinische Versorgung zu gewährleisten.
Eine unzureichende Gebrauchstauglichkeit dieser Medizinprodukte kann oftmals schwerwiegende Konsequenzen mit sich tragen, Anwenderfehler können zu schlimmen Verletzungen und – im WorstCase – gar zum Tode führen.
Medizinprodukte sollten uns eine Hilfestellung geben, die Lebensqualität zu verbessern, Untersuchungsprozesse zu optimieren und die bestmögliche medizinische Versorgung zu gewährleisten.
Daher haben wir es uns zur persönlichen Aufgabe gemacht, gemeinsam mit Herstellern eine individuelle - auf das Medizinprodukt angepasste - Analyse der Gebrauchstauglichkeit zu definieren, zu dokumentieren und den Produktlebenszyklus entsprechend aktuell zu halten. Obgleich Sie sich als Hersteller aktuell in der Ideen-, Entwicklungs- oder Zulassungsphase befinden oder Ihr Produkt vielleicht sogar bereits am Markt in Verwendung ist – kontaktieren Sie uns.
Die aktuelle Gebrauchstauglichkeits-Norm - EN 62366-1-2015+A1:2020 - steht in enger Verbindung mit dem Risikomanagement, was eine Betrachtung möglicher Anwenderfehler und resultierender Gefährdungssituationen/Gefährdungen unerlässlich macht. Anwendungsmöglichkeiten müssen - ausgehend vom bestimmungsgemäßen Gebrauch - betrachtet und analysiert werden. Der abnormale Gebrauch, wie bspw. vorsätzlich falsche Verwendung ist nicht mehr Teil der Betrachtung der Gebrauchstauglichkeit laut Norm.
Wir unterstützten Sie gerne, um den Anforderungen der aktuellen Gebrauchstauglichkeitsnorm und den entsprechenden, die Gebrauchstauglichkeit betreffenden Anforderungen aus Annex I (Grundlegende Sicherheits- und Leistungsanforderungen) der MDR (engl.: Medical Device Regulation) gerecht zu werden.